Broadly, genetic disorders may be grouped into two categories – Mendelian disorders and Chromosomal disorders. Mendelian disorders are mainly determined by alteration or mutation in the single gene. These disorders are transmitted to the offspring on the same lines as we have studied in the principle of inheritance. The pattern of inheritance of such Mendelian disorders can be traced in a family by the pedigree analysis. Most common and prevalent Mendelian disorders are Haemophilia, Cystic fibrosis, Sickle-cell anaemia, Colour blindness, Phenylketonuria, Thalassemia, etc. It is important to mention here that such Mendelian disorders may be dominant or recessive. By pedigree analysis one can easily understand whether the trait in question is dominant or recessive. Similarly, the trait may also be linked to the sex chromosome as in case of haemophilia. It is evident that this X-linked recessive trait shows transmission from carrier female to male progeny. A representative pedigree is shown in Figure 5.14 for dominant and recessive traits. Discuss with your teacher and design pedigrees for characters linked to both autosomes and sex chromosome.

(a) (b)
Figure 5.14 Representative pedigree analysis of (a) Autosomal dominant trait (for example: Myotonic dystrophy) (b) Autosomal recessive trait (for example: Sickle-cell anaemia)
Colour Blidness : It is a sex-linked recessive disorder due to defect in either red or green cone of eye resulting in failure to discriminate between red and green colour. This defect is due to mutation in certain genes present in the X chromosome. It occurs in about 8 per cent of males and only about 0.4 per cent of females. This is because the genes that lead to red-green colour blindness are on the X chromosome. Males have only one X chromosome and females have two. The son of a woman who carries the gene has a 50 per cent chance of being colour blind. The mother is not herself colour blind because the gene is recessive. That means that its effect is suppressed by her matching dominant normal gene. A daughter will not normally be colour blind, unless her mother is a carrier and her father is colour blind.
Haemophilia : This sex linked recessive disease, which shows its transmission from unaffected carrier female to some of the male progeny has been widely studied. In this disease, a single protein that is a part of the cascade of proteins involved in the clotting of blood is affected. Due to this, in an affected individual a simple cut will result in non-stop bleeding. The heterozygous female (carrier) for haemophilia may transmit the disease to sons. The possibility of a female becoming a haemophilic is extremely rare because mother of such a female has to be at least carrier and the father should be haemophilic (unviable in the later stage of life). The family pedigree of Queen Victoria shows a number of haemophilic descendents as she was a carrier of the disease.
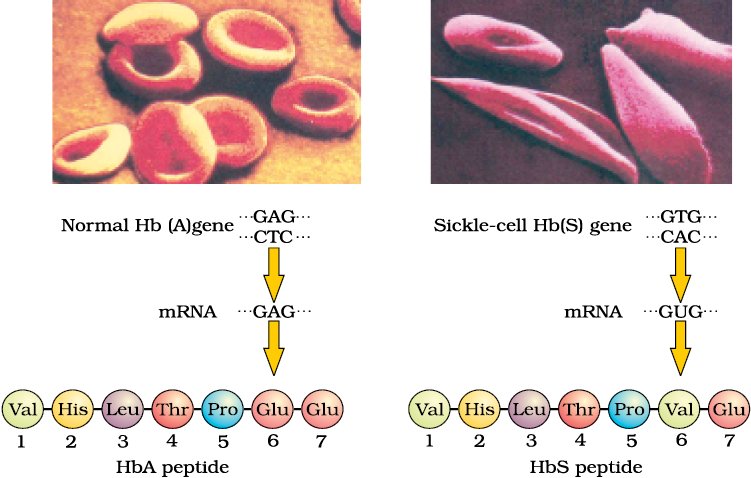
Figure 5.15 Micrograph of the red blood cells and the amino acid composition of the relevant portion of β-chain of haemoglobin: (a) From a normal individual; (b) From an individual with sickle-cell anaemia
Sickle-cell anaemia : This is an autosome linked recessive trait that can be transmitted from parents to the offspring when both the partners are carrier for the gene (or heterozygous). The disease is controlled by a single pair of allele, HbA and HbS. Out of the three possible genotypes only homozygous individuals for HbS (HbSHbS) show the diseased phenotype. Heterozygous (HbAHbS) individuals appear apparently unaffected but they are carrier of the disease as there is 50 per cent probability of transmission of the mutant gene to the progeny, thus exhibiting sickle-cell trait
(Figure 5.15). The defect is caused by the substitution of Glutamic acid (Glu) by Valine (Val) at the sixth position of the beta globin chain of the haemoglobin molecule. The substitution of amino acid in the globin protein results due to the single base substitution at the sixth codon of the beta globin gene from GAG to GUG. The mutant haemoglobin molecule undergoes polymerisation under low oxygen tension causing the change in the shape of the RBC from biconcave disc to elongated sickle like structure (Figure 5.15).
Phenylketonuria : This inborn error of metabolism is also inherited as the autosomal recessive trait. The affected individual lacks an enzyme that converts the amino acid phenylalanine into tyrosine. As a result of this phenylalanine is accumulated and converted into phenylpyruvic acid and other derivatives. Accumulation of these in brain results in mental retardation. These are also excreted through urine because of its poor absorption by kidney.
Thalassemia : This is also an autosome-linked recessive blood disease transmitted from parents to the offspring when both the partners are unaffected carrier for the gene (or heterozygous). The defect could be due to either mutation or deletion which ultimately results in reduced rate of synthesis of one of the globin chains (a and b chains) that make up haemoglobin. This causes the formation of abnormal haemoglobin molecules resulting into anaemia which is characteristic of the disease. Thalassemia can be classified according to which chain of the haemoglobin molecule is affected. In a Thalassemia, production of a globin chain is affected while in b Thalassemia, production of b globin chain is affected. a Thalassemia is controlled by two closely linked genes HBA1 and HBA2 on chromosome 16 of each parent and it is observed due to mutation or deletion of one or more of the four genes. The more genes affected, the less alpha globin molecules produced. While b Thalassemia is controlled by a single gene HBB on chromosome 11 of each parent and occurs due to mutation of one or both the genes. Thalassemia differs from sickle-cell anaemia in that the former is a quantitative problem of synthesising too few globin molecules while the latter is a qualitative problem of synthesising an incorrectly functioning globin.
© 2026 GoodEd Technologies Pvt. Ltd.

