The reactions of haloalkanes may be divided into the following categories:
1. Nucleophilic substitution
2. Elimination reactions
3. Reaction with metals.
10.7.1 Reactions of Haloalkanes
(1) Nucleophilic substitution reactions
You have learnt in Class XI that nucleophiles are electron rich species. Therefore, they attack at that part of the substrate molecule which is electron deficient. The reaction in which a nucleophile replaces already existing nucleophile in a molecule is called nucleophilic substitution reaction. Haloalkanes are substrate in these reactions. In this type of reaction, a nucleophile reacts with haloalkane (the substrate) having a partial positive charge on the carbon atom bonded to halogen. A substitution reaction takes place and halogen atom, called leaving group departs as halide ion. Since the substitution reaction is initiated by a nucleophile, it is called nucleophilic substitution reaction.
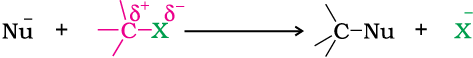
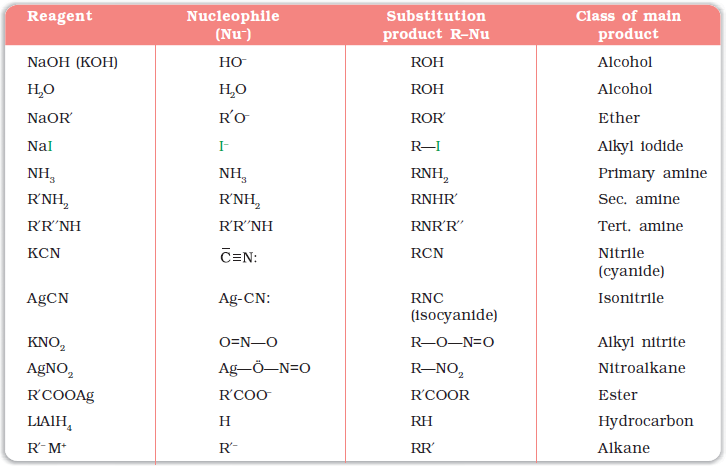
It is one of the most useful classes of organic reactions of alkyl halides in which halogen is bonded to sp3 hybridised carbon. The products formed by the reaction of haloalkanes with some common nucleophiles are given in Table 10.4.
Table 10.4: Nucleophilic Substitution of Alkyl Halides (R–X)
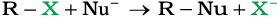
Groups like cyanides and nitrites possess two nucleophilic centres and are called ambident nucleophiles. Actually cyanide group is a hybrid of two contributing structures and therefore can act as a nucleophile in two different ways [VC≡N ↔ :C=NV], i.e., linking through carbon atom resulting in alkyl cyanides and through nitrogen atom leading to isocyanides. Similarly nitrite ion also represents an ambident nucleophile with two different points of linkage [–O—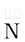 =O]. The linkage through oxygen results in alkyl nitrites while through nitrogen atom, it leads to nitroalkanes.
=O]. The linkage through oxygen results in alkyl nitrites while through nitrogen atom, it leads to nitroalkanes.
Example 10.5
Haloalkanes react with KCN to form alkyl cyanides as main product while AgCN forms isocyanides as the chief product. Explain.
Solution
KCN is predominantly ionic and provides cyanide ions in solution. Although both carbon and nitrogen atoms are in a position to donate electron pairs, the attack takes place mainly through carbon atom and not through nitrogen atom since C—C bond is more stable than C—N bond. However, AgCN is mainly covalent in nature and nitrogen is free to donate electron pair forming isocyanide as the main product.
Mechanism: This reaction has been found to proceed by two different mechanims which are described below:
(a) Substitution nucleophilic bimolecular (SN2)
The reaction between CH3Cl and hydroxide ion to yield methanol and chloride ion follows a second order kinetics, i.e., the rate depends upon the concentration of both the reactants.
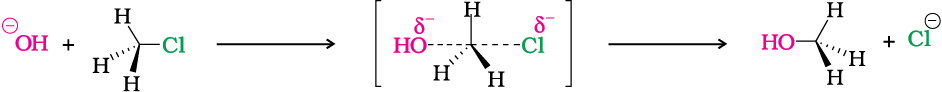
You have already learnt in Section 12.3.2 of Class XI, the solid wedge represents the bond coming out of the paper, dashed line going down the paper and a straight line representing bond in the plane of the paper.
The above reaction can be represented diagrammatically as shown in Fig. 10.2.
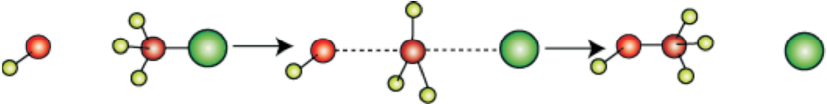
In the year 1937, Edward Davies Hughes and Sir Christopher Ingold proposed a mechanism for an SN2 reaction.
It depicts a bimolecular nucleophilic substitution (SN2) reaction; the incoming nucleophile interacts with alkyl halide causing the carbon-halide bond to break and a new bond is formed between carbon and attacking nucleophile. Here it is C-O bond formed between C and -OH. These two processes take place simultaneously in a single step and no intermediate is formed. As the reaction progresses and the bond between the incoming nucleophile and the carbon atom starts forming, the bond between carbon atom and leaving group weakens. As this happens, the three carbon-hydrogen bonds of the substrate start moving away from the attacking nucleophile. In transition state all the three C-H bonds are in the same plane and the attacking and leaving nucleophiles are partially attached to the carbon. As the attacking nucleophile closer to the carbon, C-H bonds still keep on moving in the same direction till the attacking nucleophile attaches to carbon and leaving group leaves the carbon. As a result configuration is inverted, the configuration (See box) of carbon atom under attack inverts in much the same way as an umbrella is turned inside out when caught in a strong wind. This process is called as inversion of configuration. In the transition state, the carbon atom is simultaneously bonded to incoming nucleophile and the outgoing leaving group. Such structures are unstable and cannot be isolated. Thus, in the transition state, carbon is simultaneously bonded to five atoms.
Spacial arrangement of functional groups around carbon is called its configuration. See the structures (A) and (B) given below carefully.
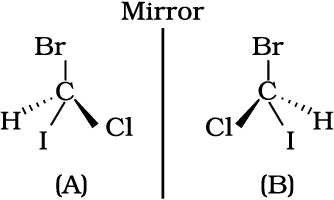
These are the two structures of the same compound. They differ in spacial arrangement of functional groups attached to carbon. Structure (A) is mirror image of Structure (B). We say configuration of carbon in structure (A) is mirror image of the configuration of carbon in structure (B).
Hughes worked under Ingold and earned a D.Sc. degree from the University of London.
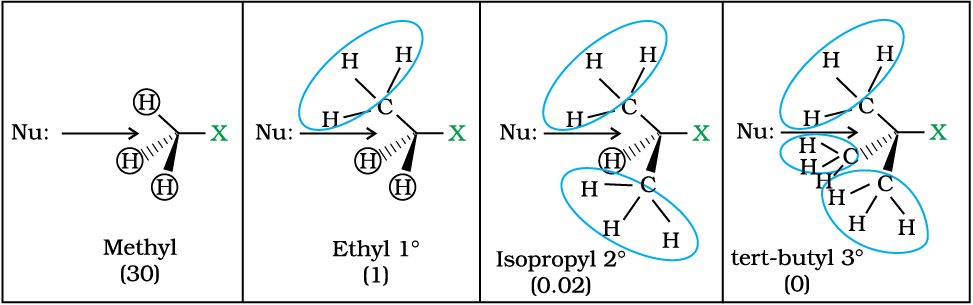
Since this reaction requires the approach of the nucleophile to the carbon bearing the leaving group, the presence of bulky substituents on or near the carbon atom have a dramatic inhibiting effect. Of the simple alkyl halides, methyl halides react most rapidly in SN2 reactions because there are only three small hydrogen atoms. Tertiary halides are the least reactive because bulky groups hinder the approaching nucleophiles. Thus the order of reactivity followed is:
Primary halide > Secondary halide > Tertiary halide.
(b) Substitution nucleophilic unimolecular (SN1)
SN1 reactions are generally carried out in polar protic solvents (like water, alcohol, acetic acid, etc.). The reaction between tert-butyl bromide and hydroxide ion yields tert-butyl alcohol and follows the first order kinetics, i.e., the rate of reaction depends upon the concentration of only one reactant, which is tert- butyl bromide.
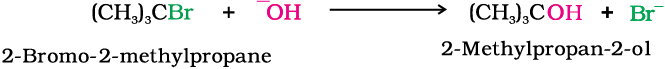
It occurs in two steps. In step I, the polarised C—Br bond undergoes slow cleavage to produce a carbocation and a bromide ion. The carbocation thus formed is then attacked by nucleophile in step II to complete the substitution reaction.
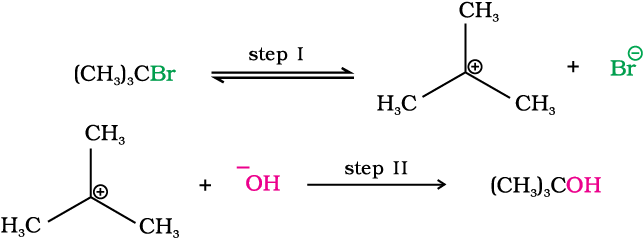
Step I is the slowest and reversible. It involves the C–Br bond breaking for which the energy is obtained through solvation of halide ion with the proton of protic solvent. Since the rate of reaction depends upon the slowest step, the rate of reaction depends only on the concentration of alkyl halide and not on the concentration of hydroxide ion. Further, greater the stability of carbocation, greater will be its ease of formation from alkyl halide and faster will be the rate of reaction. In case of alkyl halides, 3° alkyl halides undergo SN1 reaction very fast because of the high stability of 3° carbocations. We can sum up the order of reactivity of alkyl halides towards SN1 and SN2 reactions as follows:
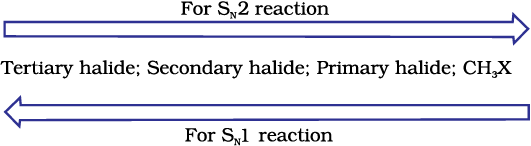
For the same reasons, allylic and benzylic halides show high reactivity towards the SN1 reaction. The carbocation thus formed gets stabilised through resonance (Unit 12, Class XI) as shown below:
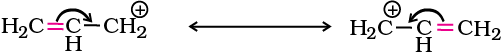
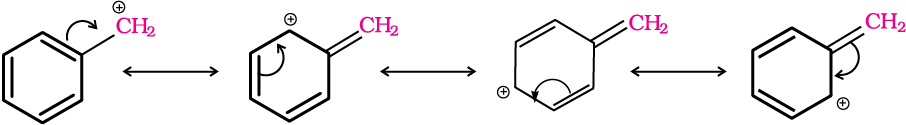
For a given alkyl group, the reactivity of the halide, R-X, follows the same order in both the mechanisms R–I> R–Br>R–Cl>>R–F.
In the following pairs of halogen compounds, which would undergo SN2 reaction faster?
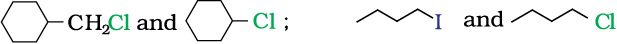
Solution
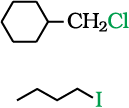 It is primary halide and therefore undergoes SN2 reaction faster.
It is primary halide and therefore undergoes SN2 reaction faster.
As iodine is a better leaving group because of its large size, it will be released at a faster rate in the presence of incoming nucleophile.
Example 10.7
Predict the order of reactivity of the following compounds in SN1 and SN2 reactions:
(i) The four isomeric bromobutanes
(ii) C6H5CH2Br, C6H5CH(C6H5)Br, C6H5CH(CH3)Br, C6H5C(CH3)(C6H5)Br
Solution
(i) CH3CH2CH2CH2Br < (CH3)2CHCH2Br < CH3CH2CH(Br)CH3 < (CH3)3CBr (SN1)
CH3CH2CH2CH2Br > (CH3)2CHCH2Br > CH3CH2CH(Br)CH3 > (CH3)3CBr (SN2)
Of the two primary bromides, the carbocation intermediate derived from (CH3)2CHCH2Br is more stable than derived from CH3CH2CH2CH2Br because of greater electron donating inductive effect of (CH3)2CH- group. Therefore, (CH3)2CHCH2Br is more reactive than CH3CH2CH2CH2Br in SN1 reactions. CH3CH2CH(Br)CH3 is a secondary bromide and (CH3)3CBr is a tertiary bromide. Hence the above order is followed in SN1. The reactivity in SN2 reactions follows the reverse order as the steric hinderance around the electrophilic carbon increases in that order.
(ii) C6H5C(CH3)(C6H5)Br > C6H5CH(C6H5)Br > C6H5CH(CH3)Br > C6H5CH2Br (SN1)
C6H5C(CH3)(C6H5)Br < C6H5CH(C6H5)Br < C6H5CH(CH3)Br < C6H5CH2Br (SN2)
Of the two secondary bromides, the carbocation intermediate obtained from C6H5CH(C6H5)Br is more stable than obtained from C6H5CH(CH3)Br because it is stabilised by two phenyl groups due to resonance. Therefore, the former bromide is more reactive than the latter in SN1 reactions. A phenyl group is bulkier than a methyl group. Therefore, C6H5CH(C6H5)Br is less reactive than C6H5CH(CH3)Br in SN2 reactions.
(c) Stereochemical aspects of nucleophilic substitution reactions
In order to understand the stereochemical aspects of substitution reactions, we need to learn some basic stereochemical principles and notations (optical activity, chirality, retention, inversion, racemisation, etc.).
(i) Optical activity: Plane of plane polarised light produced by passing ordinary light through Nicol prism is rotated when it is passed through the solutions of certain compounds. Such compounds are called optically active compounds. The angle by which the plane polarised light is rotated is measured by an instrument called polarimeter. If the compound rotates the plane of plane polarised light to the right, i.e., clockwise direction, it is called dextrorotatory (Greek for right rotating) or the d-form and is indicated by placing a positive (+) sign before the degree of rotation. If the light is rotated towards left (anticlockwise direction), the compound is said to be laevo-rotatory or the l-form and a negative (–) sign is placed before the degree of rotation. Such (+) and (–) isomers of a compound are called optical isomers and the phenomenon is termed as optical isomerism.
William Nicol (1768-1851) developed the first prism that produced plane polarised light.
(ii) Molecular asymmetry, chirality and enantiomers: The observation of Louis Pasteur (1848) that crystals of certain compounds exist in the form of mirror images laid the foundation of modern stereochemistry. He demonstrated that aqueous solutions of both types of crystals showed optical rotation, equal in magnitude (for solution of equal concentration) but opposite in direction. He believed that this difference in optical activity was associated with the three dimensional arrangements of atoms in the molecules (configurations) of two types of crystals. Dutch scientist, J. Van’t Hoff and French scientist, C. Le Bel in the same year (1874), independently argued that the spatial arrangement of four groups (valencies) around a central carbon is tetrahedral and if all the substituents attached to that carbon are different, the mirror image of the molecule is not superimposed (overlapped) on the molecule; such a carbon is called asymmetric carbon or stereocentre . The resulting molecule would lack symmetry and is referred to as asymmetric molecule. The asymmetry of the molecule along with non superimposability of mirror images is responsible for the optical activity in such organic compounds.
Jacobus Hendricus Van’t Hoff (1852-1911) received the first Nobel Prize in Chemistry in 1901 for his work on solutions.
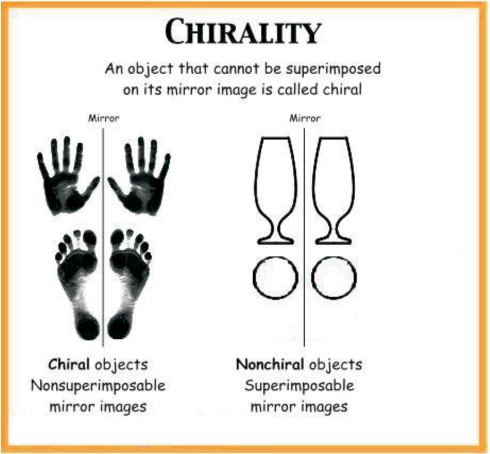
The symmetry and asymmetry are also observed in many day to day objects: a sphere, a cube, a cone, are all identical to their mirror images and can be superimposed. However, many objects are non superimposable on their mirror images. For example, your left and right hand look similar but if you put your left hand on your right hand by moving them in the same plane, they do not coincide. The objects which are non-superimposable on their mirror image (like a pair of hands) are said to be chiral and this property is known as chirality . Chiral molecules are optically active, while the objects, which are, superimposable on their mirror images are called achiral. These molecules are optically inactive.
The above test of molecular chirality can be applied to organic molecules by constructing models and its mirror images or by drawing three dimensional structures and attempting to superimpose them in our minds. There are other aids, however, that can assist us in recognising chiral molecules. One such aid is the presence of a single asymmetric carbon atom. Let us consider two simple molecules propan-2-ol (Fig.10.5) and butan-2-ol (Fig.10.6) and their mirror images.
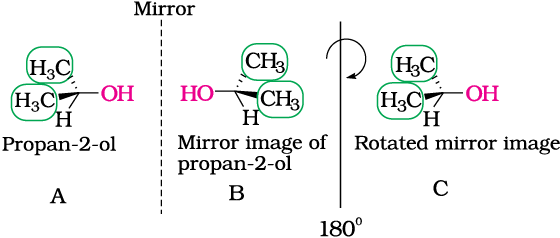
Fig 10.5: B is mirror image of A; B is rotated by 180o and C is obtained; C is superimposable on A.
As you can see very clearly, propan-2-ol (A) does not contain an asymmetric carbon, as all the four groups attached to the tetrahedral carbon are not different. We rotate the mirror image (B) of the molecule by 180° (structure C) and try to overlap the structure (C) with the structure (A), these structures completely overlap. Thus propan-2-ol is an achiral molecule.
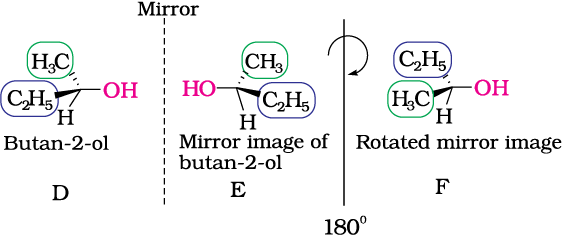
Fig 10.6: E is mirror image of D; E is rotated by 180° to get F and F is non superimposable on its mirror image D.
Butan-2-ol has four different groups attached to the tetrahedral carbon and as expected is chiral. Some common examples of chiral molecules such as 2-chlorobutane, 2, 3-dihyroxypropanal, (OHC–CHOH–CH2OH), bromochloro-iodomethane (BrClCHI), 2-bromopropanoic acid (H3C–CHBr–COOH), etc.
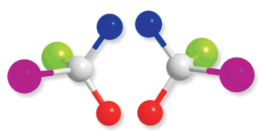
Fig. 10.7: A chiral molecule and its mirror image
The stereoisomers related to each other as non-superimposable mirror images are called enantiomers (Fig. 10.7). A and B in Fig. 10.5 and D and E in Fig. 10.6 are enantiomers.
Enantiomers possess identical physical properties namely, melting point, boiling point, refractive index, etc. They only differ with respect to the rotation of plane polarised light. If one of the enantiomer is dextro rotatory, the other will be laevo rotatory.
A mixture containing two enantiomers in equal proportions will have zero optical rotation, as the rotation due to one isomer will be cancelled by the rotation due to the other isomer. Such a mixture is known as racemic mixture or racemic modification. A racemic mixture is represented by prefixing dl or (±) before the name, for example (±) butan-2-ol. The process of conversion of enantiomer into a racemic mixture is known as racemisation.
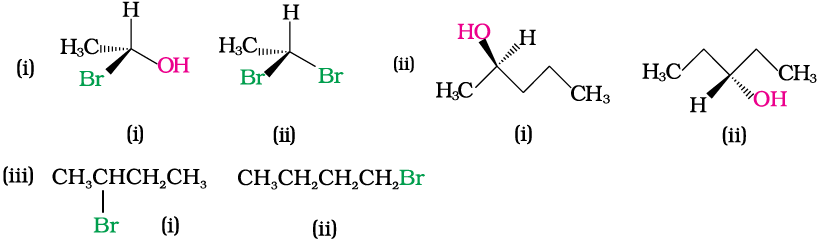
Example 10.8
Identify chiral and achiral molecules in each of the following pair of compounds. (Wedge and Dash representations according to Class XI, Fig. 12.1).
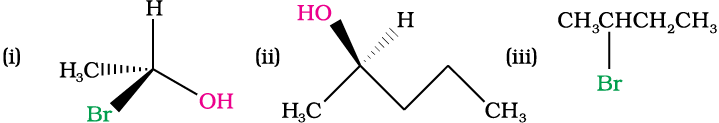
(iii) Retention: Retention of configuration is the preservation of the spatial arrangement of bonds to an asymmetric centre during a chemical reaction or transformation.
In general, if during a reaction, no bond to the stereocentre is broken, the product will have the same general configuration of groups around the stereocentre as that of reactant. Such a reaction is said to proceed with retention of the configuration. Consider as an example, the reaction that takes place when (–)-2-methylbutan-1-ol is heated with concentrated hydrochloric acid.
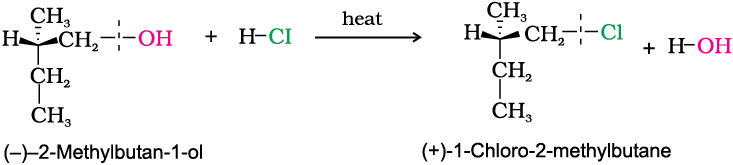
It is important to note that configuration at a symmetric centre in the reactant and product is same but the sign of optical rotation has changed in the product. This is so because two different compounds with same configuration at asymmetric centre may have different optical rotation. One may be dextrorotatory (plus sign of optical rotation) while other may be laevorotatory (negative sign of optical rotation).
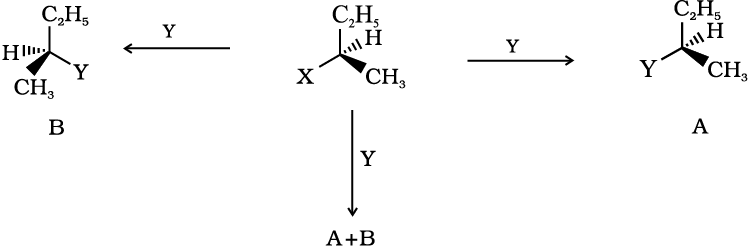
If (A) is the only compound obtained, the process is called retention of configuration. Note that configuration has been rotated in A.
If (B) is the only compound obtained, the process is called inversion of configuration. Configuration has been inverted in B.
If a 50:50 mixture of A and B is obtained then the process is called racemisation and the product is optically inactive, as one isomer will rotate the plane polarised light in the direction opposite to another.
Now let us have a fresh look at SN1 and SN2 mechanisms by taking examples of optically active alkyl halides.
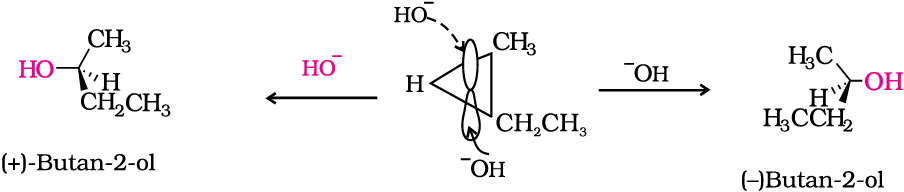
In case of optically active alkyl halides, the product formed as a result of SN2 mechanism has the inverted configuration as compared to the reactant. This is because the nucleophile attaches itself on the side opposite to the one where the halogen atom is present. When
(–)-2-bromooctane is allowed to react with sodium hydroxide, (+)-octan-2-ol is formed with the –OH group occupying the position opposite to what bromide had occupied.
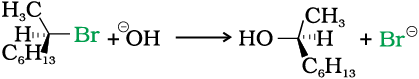
In case of optically active alkyl halides, SN1 reactions are accompanied by racemisation. Can you think of the reason why it happens? Actually the carbocation formed in the slow step being sp2 hybridised is planar (achiral). The attack of the nucleophile may be accomplished from either side of the plane of carbocation resulting in a mixture of products, one having the same configuration (the –OH attaching on the same position as halide ion) and the other having opposite configuration (the –OH attaching on the side opposite to halide ion). This may be illustrated by hydrolysis of optically active 2-bromobutane, which results in the formation of (±)-butan-2-ol.
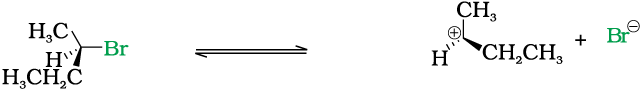
2. Elimination reactions
When a haloalkane with β-hydrogen atom is heated with alcoholic solution of potassium hydroxide, there is elimination of hydrogen atom from β-carbon and a halogen atom from the α-carbon atom.
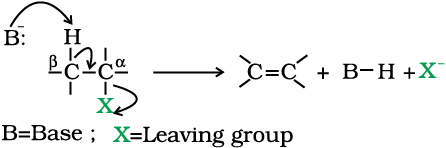
As a result, an alkene is formed as a product. Since β-hydrogen atom is involved in elimination, it is often called β-elimination.
Location of α and β carbon in a molecule
Carbon on which halogen atom is directly attached is called α-carbon and the carbon atom adjacent to this carbon is called β-carbon.
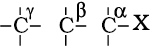
If there is possibility of formation of more than one alkene due to the availability of more than one β-hydrogen atoms, usually one alkene is formed as the major product. These form part of a pattern first observed by Russian chemist, Alexander Zaitsev (also pronounced as Saytzeff) who in 1875 formulated a rule which can be summarised as “in dehydrohalogenation reactions, the preferred product is that alkene which has the greater number of alkyl groups attached to the doubly bonded carbon atoms.” Thus, 2-bromopentane gives pent-2-ene as the major product.
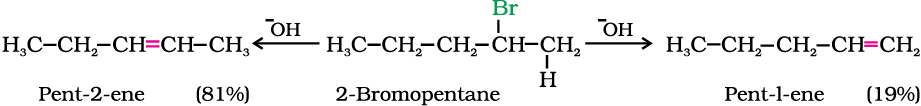
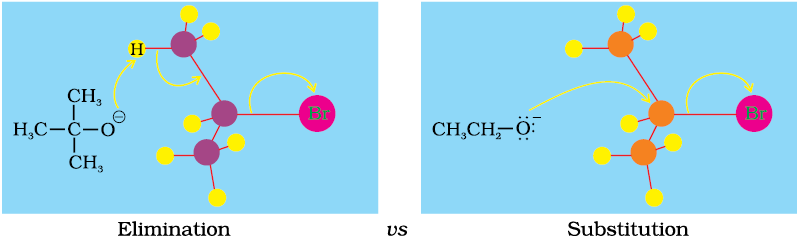
A chemical reaction is the result of competition; it is a race that is won by the fastest runner. A collection of molecules tend to do, by and large, what is easiest for them. An alkyl halide with β-hydrogen atoms when reacted with a base or a nucleophile has two competing routes: substitution (SN1 and SN2) and elimination. Which route will be taken up depends upon the nature of alkyl halide, strength and size of base/nucleophile and reaction conditions. Thus, a bulkier nucleophile will prefer to act as a base and abstracts a proton rather than approach a tetravalent carbon atom (steric reasons) and vice versa. Similarly, a primary alkyl halide will prefer a SN2 reaction, a secondary halide- SN2 or elimination depending upon the strength of base/nucleophile and a tertiary halide- SN1 or elimination depending upon the stability of carbocation or the more substituted alkene.
3. Reaction with metals
Most organic chlorides, bromides and iodides react with certain metals to give compounds containing carbon-metal bonds. Such compounds are known as organo-metallic compounds. An important class of organo-metallic compounds discovered by Victor Grignard in 1900 is alkyl magnesium halide, RMgX, referred as Grignard Reagents. These reagents are obtained by the reaction of haloalkanes with magnesium metal in dry ether.


Victor Grignard had a strange start in academic life for a chemist - he took a maths degree. When he eventually switched to chemistry, it was not to the mathematical province of physical chemistry but to organic chemistry. While attempting to find an efficient catalyst for the process of methylation, he noted that Zn in diethyl ether had been used for this purpose and wondered whether the Mg/ether combination might be successful. Grignard reagents were first reported in 1900 and Grignard used this work for his doctoral thesis in 1901. In 1910, Grignard obtained a professorship at the University of Nancy and in 1912, he was awarded the Nobel prize for Chemistry which he shared with Paul Sabatier who had made advances in nickel catalysed hydrogenation.
In the Grignard reagent, the carbon-magnesium bond is covalent but highly polar, with carbon pulling electrons from electropositive magnesium; the magnesium halogen bond is essentially ionic.
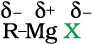
Grignard reagents are highly reactive and react with any source of proton to give hydrocarbons. Even water, alcohols, amines are sufficiently acidic to convert them to corresponding hydrocarbons.

Wurtz reaction
Alkyl halides react with sodium in dry ether to give hydrocarbons containing double the number of carbon atoms present in the halide. This reaction is known as Wurtz reaction (Unit 13, Class XI).

© 2026 GoodEd Technologies Pvt. Ltd.

